Ein problematisches Bild
Laut einer Studie aus dem Jahr 2001 [1] sind 9,4 % der Frauen über vierzig Jahre (also bereits sehr jung) und 11,8 % der Männer über sechzig Jahre von Osteoporose betroffen. Die Zahlen steigen beträchtlich an, wenn es sich um Osteopenie handelt (47,2 % für Frauen und 46,1 % für Männer bei denselben Bevölkerungsgruppen), und zwar in noch deutlicherer Weise im Verhältnis zum Alter der betrachteten Patientengruppe. Diese Daten weisen auf ein wesentliches Problem hin, das übrigens nicht einfach zu lösen ist.
Die Leitlinien [2,3] stützen sich bei der Behandlung dieser Erkrankung vor allem auf die Einnahme von Kalzium und Vitamin D, Östrogenen und Bisphosphonaten (bei Glukokortikoid-induzierter Osteoporose), konzentrieren sich aber vor allem auf die Prävention in jüngerem Alter. Leider ist es nicht ungewöhnlich, dass diese therapeutischen Strategien nicht die gewünschten Ergebnisse erzielen, hauptsächlich aufgrund von endokrin-metabolischen Situationen, die ihre Anwendbarkeit einschränken und sogar zu einer Reihe von Nebenwirkungen wegen der Therapien selbst führen können.
Um die effektivste therapeutische Strategie zu finden, ist es zunächst notwendig, die Hauptgründe für die fortschreitende Knochendemineralisation zu verstehen, da diese unterschiedlich sein können und nicht unbedingt durch eine einzige Therapie gelöst werden können.
Den Knochenstoffwechsel verstehen
Die Knochenstruktur [4-7] besteht aus hochmineralisiertem Bindegewebe (die Mineralsubstanz macht ca. 70% des Knochens aus) und wird von den notwendigen Blut- und Lymphgefäßen durchzogen. Zu der im Knochen vorhandenen organischen Substanz, die größtenteils aus Kollagen Typ 1 besteht, gehören auch Knochenzellen, die an den verschiedenen Phasen des Knochenstoffwechsels beteiligt sind. Diese Zellen lassen sich in zwei grundlegende Kategorien einteilen:
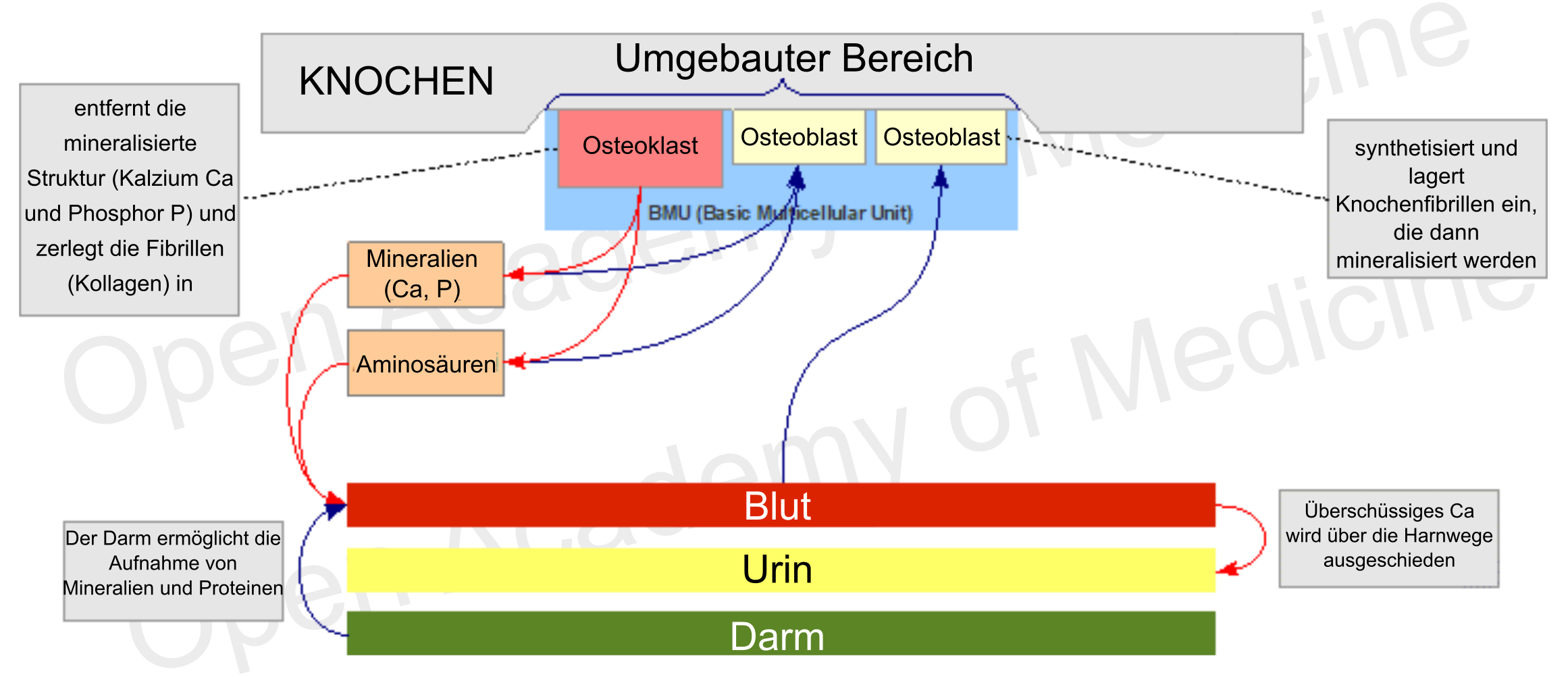
Abbildung 1: Wichtigste Wechselwirkungen im Knochenstoffwechsel
Diese Zellen agieren nicht unabhängig voneinander, sondern bewegen sich in Gruppen, die als BMUs (Basic Multicellular Units, multizelluläre Grundeinheiten) bezeichnet werden und im Wesentlichen aus einer Front von Osteoklasten bestehen, auf die eine bestimmte Anzahl von Osteoblasten folgt. Das Vorherrschen der Aktivität einer dieser beiden Zelltypen ist der Hauptfaktor für die Differenzierung der Wachstums-, Erhaltungs- (Umbau-) und Demineralisationsphase des Skeletts. Die höchste Knochendichte wird in der Regel um das zwanzigste Lebensjahr erreicht, während sie nach dem dreißigsten Lebensjahr normalerweise zu sinken beginnt.

Abbildung 2: Schematische Darstellung der Knochenzellen-Balance
Das Gleichgewicht dieser antagonistischen Prozesse hängt von mehreren Faktoren ab, die den Zeitpunkt der Zellapoptose (die Lebensdauer von Osteoklasten ist im Allgemeinen viel kürzer als die von Osteoblasten) bzw. die Entstehung von Osteoblasten und Osteoklasten beeinflussen können.

Abbildung 3: BMU-Aktivierung
Während ihrer Knochenumbauaktivität können Osteoblasten in der von ihnen gebildeten Matrix stecken bleiben und sich zu Osteozyten entwickeln: Das sind Zellen, die durch lange zytoplasmatische Verzweigungen ein durch das Knochengewebe verlaufendes Austauschnetz bilden, das sowohl mit dem Knochenmark als auch mit den BMUs kommunizieren kann (z.B. um den Umbau eines bestimmten Knochenfragments zu stimulieren).
Das nicht umgebaute Knochengewebe ist im Allgemeinen durch eine Mikroschicht aus Kollagen geschützt, die von einer Schicht aus Knochenauskleidungszellen (sogenannte Lining Cells, die von inaktivierten Osteoblasten abstammen) umgeben ist: Dieses organische Material verhindert das Anhaften von Osteoklasten an der mineralisierten Oberfläche und wird möglicherweise von den Auskleidungszellen entfernt, die durch die Vermittlung von Osteozyten zur Produktion von Kollagenase (die Kollagen Typ 1 und 2 abbaut) angeregt werden können.
Änderungen bei der Knochendichte
Die Abnahme der Knochendichte ist ein Prozess, der zwischen dem dreißigsten und vierzigsten Lebensjahr sowohl bei Männern als auch bei Frauen beginnt, und zwar aufgrund eines zunehmenden Missverhältnisses bei den Prozessen im Rahmen der Knochengewebe-Regeneration, die durch eine höhere osteoklastische Aktivität gekennzeichnet sind. In den meisten Fällen hängt diese Entwicklung nicht mit einer niedrigen Kalziumzufuhr zusammen, sondern mit anderen, vor allem endokrinen und metabolischen Faktoren, die die Aktivität von Osteoblasten und Osteoklasten beeinflussen können. Es ist jedoch das Überwiegen der Osteoklasten gegenüber den Osteoblasten, das zu einer allmählichen Knochendemineralisation (und folglich zu Knochenbrüchigkeit) führt.
Endokrine Faktoren
Zu den Faktoren, die am häufigsten an dem Ungleichgewicht zwischen der Wirkung von Osteoklasten und Osteoblasten beteiligt sind, gehört - neben dem Mangel an Sexualhormonen (Östrogen, Testosteron) - der chronische Überschuss an Glucocorticoiden [8-15] (sowohl endogen als auch exogen; BIA-ACC - Flat Low/High HPA axis index).

Abbildung 4: Glucocorticoide und Knochenstoffwechsel (BIA-ACC - Flat Low/High HPA axis index)
Die Auswirkungen dieses Überschusses auf den Knochenstoffwechsel sind nachgewiesen, und zwar durch Mechanismen, die sowohl direkt auf die Knochenzellen wirken als auch durch lokale und systemische Wechselwirkungen mit Hormonen, Wachstumsfaktoren und Zytokinen vermittelt werden.

Abbildung 5: Glucocorticoide und RANKL/OPG/RANK-Bindungen
Auf endokriner Ebene reduzieren Glucocorticoide die intestinale Absorption von Kalzium, während sie gleichzeitig dessen renale Ausscheidung erhöhen und so eine Hypokalzämie verursachen. Dieser Zustand ist mit einer erhöhten Aktivierung von Osteoklasten und damit mit einer erhöhten Knochenresorption verbunden (ob dies auf einen Glucocorticoid-induzierten Hyperparathyreoidismus oder auf eine erhöhte PTH-Empfindlichkeit der Osteoblasten, welche auf dieses Hormon mit einer Aktivierung der Osteoklasten reagieren würden, zurückzuführen ist, muss noch durch Untersuchungen geklärt werden). Glucocorticoide hemmen auch die GH-Sekretion und die Sexualhormonsynthese, während der Anstieg der Glucocorticoide mit einer erhöhten Kollagenase-Expression einhergeht, was sich negativ auf die organische Knochensubstanz auswirkt (siehe HPA-Achse für weitere Details: Schlaflosigkeit, Stimmungsstörungen, Angstzustände, Melancholie, Depression, MUS, Panikattacken).
Glucocorticoide haben auch direkte Auswirkungen auf Knochenzellen über das RANKL/OPG-Zytokin-System [7,16], indem sie die Expression des ersteren erhöhen und die des letzteren verringern und somit die Bindung von RANKL-Zytokinen (die auf den Membranen von Osteoblasten exprimiert werden) an RANK-Rezeptoren (auf den Membranen von Osteoklastenvorläufern) fördern; diese Bindung stimuliert die Differenzierung und Aktivierung von Osteoklasten und hemmt deren Apoptose (programmierter Zelltod).
Der Anstieg der Glucocorticoide hemmt auch den Osteoblasten-Differenzierungsfaktor, wodurch die Knochenregenerationsaktivität reduziert wird, und erhöht auch die Apoptose von Osteoblasten und Osteozyten. Die Osteozyten-Apoptose spielt ebenfalls bei der Knochenbrüchigkeit eine grundlegende Rolle, da Osteozyten - wie bereits gesehen - an der BMU-Aktivierung und den Knochenumbauprozessen beteiligt sind.
Die beschriebenen Effekte können durch einen Überschuss sowohl an exogenen Glucocorticoiden [9,14] (und sollten daher bei Patienten, die Glucocorticoide zu therapeutischen Zwecken einnehmen, besonders beachtet werden) als auch an endogenen Glucocorticoiden beobachtet werden. Zu den Faktoren, die an einem chronischen Cortisol-Überschuss und an der damit verbundenen Verschiebung des zirkadianen Rhythmus (BIA-ACC - Flat Low/High HPA axis index) beteiligt sind, gehören das Vorliegen chronischer Entzündungsprozesse, chronischer Stress auf die HPA-Achse und schlechte Ernährung [26-29].
Hoch titrierte EPA+DHA-Omega-3-Fettsäuren reduzieren nachweislich die Expression von RANKL-Zytokinen und erhöhen die OPGs [17-21], wodurch die Aktivierung von Osteoklasten verringert wird; die hoch titrierte EPA+DHA-Omega-3-Aufnahme ist auch mit einer Verringerung der Konzentration proinflammatorischer Zytokine (z. B. IL-6 und TNF-α) verbunden, so dass die Wirkungen dieser Fettsäuren in mehrere Richtungen zur Verringerung der Knochenresorption beitragen. Neuere Studien [22,23] haben auch berichtet, dass bestimmte Hopfenflavonoide (Humulus Lupulus L.), insbesondere Xanthohumol, in der Lage sind, selektiv mit Östrogenrezeptoren zu interagieren und eine Östrogen-ähnliche Aktivität auf den Knochenstoffwechsel auszuüben (und damit die Demineralisation zu reduzieren), ohne die Nebenwirkungen von Hormontherapien hervorzurufen (mehrere Publikationen berichten über ein erhöhtes Risiko für Brust- und Gebärmutterkrebs), während sie auch eine schützende Wirkung gegen diese Probleme zeigen (siehe hoch titrierte Nahrungsergänzung mit EPA+DHA+Humulus Lupulus L.).
Metabolische Faktoren

Abbildung 6: Azidose und Knochenstoffwechsel (siehe Supplementierung mit Puffersystemen)
Der Knochenumbau ist nicht nur maßgeblich an der Geweberegeneration und an der Anpassung der Skelettstruktur an die mechanischen Belastungen, denen sie ausgesetzt ist, beteiligt, er ist auch ein wesentlicher Prozess für die Kalziumhomöostase, da das Knochengewebe das wichtigste Kalziumreservoir des Körpers ist. Das Gleichgewicht mit dem freien ionisierten Kalzium im Plasma (und sein möglicher Mangel) ist jedoch nicht der Hauptaspekt, der unter diesem Gesichtspunkt zu betrachten ist: Es ist nämlich bekannt, dass das Knochengewebe eine grundlegende Rolle beim Ausgleich des Körper-pH spielt, da es selbst ein Puffersystem gegen Azidose ist [24,25]. Die zur Erhaltung des Körper-pH erzeugte Entkalkung kann offensichtlich zu einer erhöhten Knochenbrüchigkeit und Hyperkalzämie führen.
Aus therapeutischer Sicht ist daher zur Bewältigung einer solchen Situation eine angemessene Supplementierung mit Phosphat- und Bikarbonat-Puffersystemen erforderlich (siehe Supplementierung mit Puffersystemen): Die Wiederherstellung des pH-Wertes hat Vorrang [30-31] gegenüber der Kalziumergänzung, da Kalzium aufgrund der fortschreitenden Knochendemineralisation bereits übermäßige Plasmakonzentrationen aufweisen kann.
Warum sollte eine Therapie eingeleitet werden?
Wenn das Knochengewebe aus einem oder mehreren der besprochenen Gründe einer Demineralisation unterliegt, muss der Patient behandelt werden, um diese Tendenz zu korrigieren, bevor die klassische Kalzium- und Vitamin-D-Supplementierung (die möglicherweise nicht notwendig ist) in Betracht gezogen wird. Der Überschuss an diesen Komponenten würde ohne die systemischen Bedingungen für die Erholung des Knochengewebes leicht zu Nebenwirkungen wie Weichteilverkalkungen oder Nierensteinen führen und wäre nicht in der Lage, den fortschreitenden Knochenmineralverlust zu begrenzen.

Abbildung 7: Nebenwirkungen einer Therapie, die nicht auf endokrine und metabolische Aspekte eingeht
Autoren: Dario Boschiero - Datum: 14/12/2020
Achtung: Die Inhalte dürfen ausschließlich für persönliche Lernzwecke frei verwendet werden. Die Nutzung wird durch Gesetz Nr. 633/1941 und darauffolgende Änderungen sowie durch das Urheber- und Patentrecht geregelt. Jegliche Nutzung zu kommerziellen und gewinnorientierten Zwecken ist verboten.
Literaturverzeichnis




